
Introduction
Genomic DNA (gDNA) contamination in RNA samples is a quality control failure that rarely announces itself. Unlike sample degradation or obvious procedural errors, gDNA contamination evades standard detection while quietly distorting RT-qPCR quantification, RNA-seq differential expression results, and transcriptomic pathway conclusions.
Research shows that even DNase-treated RNA can harbour approximately 1.8% residual gDNA — a level invisible on standard gel electrophoresis, yet enough to generate false differentially expressed genes and spurious pathway enrichment results.
Reliable RNA data starts before sequencing platform selection or statistical modelling. It starts with understanding how gDNA enters your samples and applying consistent assessment and removal protocols before contamination reaches your assay.
Key Takeaways
- gDNA contamination originates from incomplete DNase digestion, aggressive mechanical lysis, or challenging sample types (FFPE, blood tubes)
- Low-abundance transcripts suffer inflated signals, generating false positives in differential expression and pathway analysis
- Assessment tools include spectrophotometry ratios, Bioanalyzer profiling, RT-PCR with intron-spanning primers, and RNA-seq intergenic mapping
- Prevention relies on in-solution DNase digestion, magnetic bead-based extraction, and library prep selection (Poly(A) vs. Ribo-Zero)
- Multi-stage QC — pre-extraction, in-process, and post-extraction — is the most reliable path to DNA-free RNA
Common Causes of gDNA Contamination in RNA Samples
While gDNA contamination manifests as a single outcome, it typically results from multiple upstream failures during sample handling, extraction, or processing. Identifying which failure mode is active in your workflow is essential to implementing an effective solution rather than applying generic troubleshooting.
Incomplete or Insufficient DNase Digestion
DNase I digestion serves as the primary enzymatic defense against gDNA carry-over, but standard on-column protocols frequently prove insufficient for complete removal. In-solution digestion—performed with doubled enzyme units and extended incubation times—delivers measurably more thorough gDNA elimination than the brief on-column treatments included in most commercial kits.
The typical failure scenario unfolds in labs following manufacturer default protocols: teams skip the optional extended digestion step, assuming that a 15-minute on-column DNase treatment will suffice. The result is residual gDNA levels invisible on agarose gels but readily detectable by RT-PCR no-RT controls or RNA-seq bioinformatic mapping analysis.
On-column access is spatially constrained compared to solution-phase digestion — a limitation that becomes critical for samples with high DNA loads where complete enzymatic contact is required.
Aggressive Mechanical Lysis Releasing Nuclear DNA
Harsh mechanical disruption methods—bead-beating, vigorous vortexing with TRIzol, or repeated freeze-thaw cycling—physically shear the nuclear envelope, releasing substantial quantities of chromosomal DNA that co-purifies alongside RNA. This problem intensifies for tissues with high nuclear content, where the ratio of nuclear DNA to cytoplasmic RNA already favours contamination.
TRIzol/phenol-chloroform protocols demonstrate approximately 142–237 times higher gDNA contamination compared to silica spin-column methods. The mechanism involves phase separation chemistry: gDNA partitions to the interphase and organic phase during TRIzol extraction, but incomplete separation or vortexing during the isopropanol precipitation step causes DNA to co-precipitate with RNA.
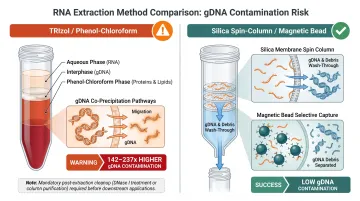
Without mandatory post-extraction spin-column cleanup, TRIzol-based methods carry unacceptable contamination risk for sequencing applications — residual phenol and co-precipitated DNA both contribute to downstream failures.
These extraction-level risks compound when the sample type itself introduces structural challenges.
Challenging Sample Types
FFPE samples present elevated gDNA contamination risk due to formalin fixation chemistry: long-term storage produces DNA fragments (150–500 bp) that overlap in size with target RNA fragments. This size similarity gives fragmented gDNA high co-extraction probability during purification steps designed to capture small RNA molecules.
Published RNA-seq data shows gDNA contamination in Ribo-Zero libraries from FFPE samples ranging from 2.2% to 7.5%, with some normal tissue samples reaching 22.7%. These contamination levels directly compromise differential expression accuracy for low-abundance genes.
Blood preservation tubes (PAXgene, Tempus) require mandatory additional spin-column cleanup post-extraction. These tubes lyse cells immediately upon collection to stabilize RNA, but this pre-lysis releases both RNA and DNA into solution simultaneously, increasing co-purification risk.
A critical anticoagulant consideration: heparin must never be used for blood samples destined for nucleic acid extraction. Heparin co-purifies with nucleic acids and inhibits both Taq DNA polymerase and reverse transcriptase, causing false negatives or inflated quantification in RT-qPCR. Use EDTA or citrate anticoagulants instead.
Library Preparation Method Amplifying Residual Contamination
Your library prep choice directly impacts gDNA contamination risk, independent of extraction quality. Ribo-Zero depletion libraries capture significantly more gDNA than Poly(A) selection libraries because Ribo-Zero retains all non-rRNA molecules—including gDNA fragments—while Poly(A) selection uses oligo-dT probes that structurally require a ~250 bp poly-A tail for capture.
Since gDNA lacks poly-A tails, Poly(A) selection provides built-in gDNA exclusion. This makes library prep method selection a contamination risk factor to consider during experimental design, not just a post-extraction decision.
Why gDNA Contamination Is More Dangerous Than It Looks
Disproportionate Impact on Low-Abundance Transcripts
gDNA generates sequencing reads that map across the entire genome (exons, introns, and intergenic regions), creating uniform low-level signal across all genomic loci. This background inflates expression levels preferentially for genes near baseline, where gDNA-derived counts make up a larger fraction of total signal.
Analysis of RNA-seq libraries showed that 94.1% of genes whose expression correlated with gDNA concentration were expressed at low levels (<0 log₂ FPKM). Highly expressed genes remain largely unaffected because the legitimate mRNA signal overwhelms the gDNA background, but weakly expressed genes become disproportionately vulnerable to false-positive differential expression calls.
Spurious Pathway Enrichment and Biological Conclusions
The downstream consequences extend beyond individual gene calls. False differentially expressed genes (DEGs) feed directly into pathway enrichment analyses (KEGG, GO, Reactome), producing spurious biological conclusions that appear statistically significant yet reflect contamination artifacts rather than genuine biology.
Research comparing contaminated versus clean libraries demonstrated:
- 1% gDNA contamination: 1,134 DEGs (14.2% directly attributable to gDNA)
- 10% gDNA contamination: 5,533 DEGs with 35 falsely enriched KEGG pathways

This risk compounds in discovery studies comparing samples with similar expression profiles, such as replicate comparisons within the same tissue type. Even small gDNA contributions can generate artifactual pathway hits that mislead biological interpretation.
Misidentification of Novel Transcribed Elements
Perhaps the most hazardous consequence involves misidentifying gDNA contamination as genuine biological signal. Studies of single-exon long non-coding RNAs (lncRNAs) demonstrated that MALAT1 was detected in 100% of plasma samples before DNase treatment but only 13.3% after treatment—revealing that 86.7% of initial "positive" results reflected gDNA contamination, not authentic MALAT1 expression.
The same pattern applies to any transcript lacking exon-intron junctions. Treat all low-abundance, intergenic calls as suspect until gDNA contamination has been explicitly ruled out — particularly for:
- Single-exon lncRNAs (NEAT1, DLEU, NORAD, KCNQ1OT1)
- Novel or unannotated intergenic transcripts
- Enhancer RNAs with no confirmed splice sites
How to Assess gDNA Contamination in Your RNA Samples
Assessment requires matching the right detection method to the right workflow stage. The following four-tier approach progresses from rapid screening to quantitative confirmation.
Spectrophotometry (Nanodrop) and Fluorometry (Qubit)
Spectrophotometry provides rapid purity indicators but cannot distinguish gDNA from RNA. The 260/280 ratio (target 1.8–2.1 for RNA) indicates protein contamination, while the 260/230 ratio (target >1.5) flags organic contaminant carryover. Any molecule absorbing at 260 nm—DNA, RNA, free nucleotides, or even some dyes—contributes to total absorbance.
Critical limitation: Spectrophotometer ratios become unreliable at concentrations approaching 10 ng/µL because trace contaminants dominate the absorbance signal at low sample concentrations. For accurate quantification, fluorometric measurement (Qubit, Quantus) is mandatory—these instruments use dyes that fluoresce only when bound to specific target molecules, providing orders of magnitude greater sensitivity and the ability to distinguish DNA from RNA.
Gel Electrophoresis and Bioanalyzer Profiling
Agarose gel electrophoresis stained with ethidium bromide or SYBR dyes reveals gDNA contamination as a high-molecular-weight smear or distinct band well above the 10 kb marker, clearly separated from the expected ribosomal RNA bands (18S and 28S) and mRNA smear in the 200–2,000 bp range.
Agilent Bioanalyzer RNA chips provide higher resolution: gDNA contamination appears as unexpected peaks or elevated baseline signal past (to the right of) the 28S ribosomal peak, typically in the higher molecular weight region. Any visible DNA band on gel or an anomalous Bioanalyzer peak in this region means the sample needs DNase re-treatment before downstream use.
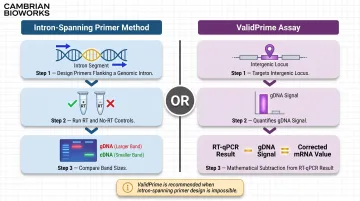
RT-PCR–Based Detection with Intron-Spanning Primers
This method offers the highest sensitivity and specificity for trace gDNA detection. Design PCR primers spanning a genomic intron: if gDNA is present, amplification from the genomic template produces a larger product (exon + intron) than the spliced mRNA-derived cDNA product (exons only). Running a no-RT control reaction (omitting reverse transcriptase) confirms gDNA as template — any amplification in the no-RT control definitively demonstrates contamination.
The ValidPrime assay extends this principle into a formalized, quantitative framework. ValidPrime targets non-transcribed intergenic loci with no known transcriptional activity, using a gDNA reference sample to mathematically estimate and subtract gDNA-derived signal from total RT-qPCR signal for any gene of interest.
ValidPrime is particularly valuable when intron-spanning primer design is impossible:
- Applies to single-exon (intronless) genes and genes with processed pseudogenes
- Corrects for gDNA contributing up to ~60% of total signal
- Reduces required control reactions by >95% compared to traditional no-RT controls
Bioinformatic Assessment in RNA-seq (Intergenic Mapping Ratio)
For RNA-seq data, gDNA contamination can be estimated post-sequencing by calculating the proportion of reads mapping to intergenic regions (genomic regions not overlapping annotated genes or transcripts). Legitimate mRNA-derived reads should map predominantly to exonic regions, while gDNA-derived reads distribute across the entire genome including intergenic space.
Regression modeling of intergenic mapping ratios estimated approximately 1.8% residual gDNA in DNase-treated total RNA, with contamination ranging from 0.7% to 22.7% across normal human tissue samples. Elevated intergenic mapping above tissue-specific baselines indicates residual contamination requiring investigation.
Ribo-Zero libraries require this check more urgently than Poly(A) selection libraries given their higher gDNA capture rate. Standard QC tools for initial assessment include:
- FastQC — rapid per-sample quality metrics and read distribution overview
- FastQ Screen — flags reads mapping to unexpected genomes or genomic backgrounds before deeper mapping analysis
How to Prevent gDNA Contamination During RNA Extraction
Prevention requires a multi-layer strategy: no single step guarantees DNA-free RNA. Instead, build protection through protocol design, extraction method selection, and consistent execution.
Prioritize In-Solution DNase Digestion Over On-Column Digestion
After initial RNA isolation, perform in-solution DNase treatment by doubling both the recommended enzyme units and incubation time compared to kit defaults. Follow with spin-column re-purification — not just washing — to physically remove digested DNA fragments and residual enzyme.
In-solution digestion gives the enzyme full three-dimensional access to gDNA, unlike on-column digestion where spatial constraints limit enzymatic contact. This matters most for samples with high DNA loads — FFPE tissue, blood preservation tubes, or any sample showing high-molecular-weight contamination on initial QC.
Apply this approach to any sample heading into RNA-seq or quantitative expression analysis.
Use Magnetic Bead-Based or Column-Based Extraction Over Organic Solvent Methods
Prefer silica spin-column or magnetic bead-based RNA extraction over TRIzol/phenol-chloroform protocols for samples proceeding to sequencing. If organic methods are required for specific tissue types, always perform post-extraction spin-column cleanup to remove co-precipitated DNA.
Column and bead methods use selective binding chemistry — silica membranes or paramagnetic beads under specific buffer conditions — that preferentially retain RNA while letting gDNA flow through or wash away. Organic methods rely on phase separation, where incomplete partitioning or mechanical disruption causes gDNA co-precipitation.
Automation strengthens this further. Platforms with pre-programmed protocols, validated wash steps, and consistent incubation times eliminate operator-dependent variability — one of the leading causes of batch-to-batch inconsistency in DNase treatment outcomes. Cambrian Bioworks' extraction systems, for example, use pre-filled cartridges with integrated DNase treatment and a closed-system design that fits inside a biosafety hood, removing manual variability from the equation entirely.
Select Library Preparation Method With Awareness of gDNA Risk
When studying mRNA from good-quality RNA, favour Poly(A) selection library prep over Ribo-Zero depletion. Poly(A) selection's oligo-dT probe design structurally excludes most gDNA, which lacks the ~250 bp poly-A tail required for capture — providing built-in gDNA filtration that Ribo-Zero workflows simply don't offer.
Reserve Ribo-Zero for non-coding RNA studies or degraded samples (such as FFPE) where Poly(A) selection is infeasible. In those cases, apply stricter upstream DNase treatment to compensate for the higher contamination risk.
Make this decision during experimental design. The library prep method directly determines how much gDNA tolerance your workflow can afford.
Maintain a DNA-Free Workspace and Prevent Cross-Contamination
Essential practices:
- Designate separate pipettes and workspaces for pre- and post-extraction steps
- Use certified RNase/DNase-free consumables exclusively
- Avoid heparin anticoagulants for blood samples (use EDTA or citrate)
- Perform thorough washes during spin-column protocols (minimum two washes plus dry spin before elution)
Environmental and reagent contamination introduces exogenous DNA that downstream DNase treatment cannot always rescue. Implement these as standing lab practices for all RNA workflows, not selective case-by-case decisions.
Long-Term Best Practices for gDNA Contamination Control
Sustained contamination control requires institutional practices, not just individual protocol adherence:
Routine QC checkpoints: Run agarose gel or Bioanalyzer analysis on a representative sample subset from every extraction batch. Any batch showing high-molecular-weight DNA signal should be flagged for mandatory re-treatment before downstream use.
Protocol documentation: Record extraction protocols with DNase enzyme lot numbers, incubation times, spin parameters, and kit expiration dates. Track these contamination indicators over time to catch protocol drift or reagent degradation early:
- Gel appearance (high-molecular-weight bands)
- 260/280 absorbance ratios
- Intergenic mapping percentages in sequencing data
Targeted training: Train all personnel on gDNA contamination risks with specific attention to highest-risk sample types—FFPE blocks, blood preservation tubes, tissues with high nuclear content. Re-train when kit formulations or instrument protocols change to ensure continued awareness.
Automation evaluation: Automated extraction platforms enforce standardized protocol execution and reduce manual pipetting variability. When evaluating platforms, prioritize built-in contamination controls such as UVC decontamination lamps, closed cartridge systems, and HEPA filtration. A compact footprint compatible with biosafety hood placement is worth considering for workflows that require containment.
Conclusion
gDNA contamination in RNA samples stems from clear, addressable root causes: insufficient DNase treatment, suboptimal extraction methods, challenging sample matrices, and library prep choices that amplify rather than filter residual DNA. Identifying which failure mode operates in your specific workflow drives an effective prevention strategy — not generic troubleshooting.
A proactive quality control approach protects data integrity and saves the time and cost of failed experiments or misleading biological conclusions. The core pillars of that approach are consistent:
- Plan extraction conditions around your sample matrix before starting
- Apply both enzymatic (DNase) and physical removal methods where contamination risk is high
- Verify RNA purity post-extraction before committing samples to downstream assays
- Choose library prep chemistries that account for residual DNA signals
Getting these steps right is what separates reproducible transcriptomic data from results that require explaining.
Frequently Asked Questions
What is the difference between gDNA and cDNA?
gDNA is the organism's full chromosomal DNA — present in every nucleated cell — including introns, regulatory regions, and intergenic sequences. cDNA is synthesized from mRNA via reverse transcription and contains only spliced, expressed sequences. gDNA contamination is a pre-analytical problem; cDNA is the intentional product of downstream RNA processing.
How to check genomic DNA contamination in cDNA?
Use intron-spanning PCR primers in a no-RT control reaction (omitting reverse transcriptase): amplification in the absence of RT confirms genomic DNA carry-over because only gDNA—not cDNA—would serve as template for the larger intron-containing amplicon. This provides definitive, sequence-specific confirmation of gDNA presence.
What happens if a DNA sample is contaminated?
gDNA contamination inflates apparent transcript levels, particularly for low-abundance genes, generating false differentially expressed genes (DEGs) and artifactual pathway enrichment results. This misdirects biological conclusions and is hardest to detect when gDNA signal constitutes a large fraction of total detected signal.
Can DNase treatment completely remove gDNA from RNA samples?
DNase treatment substantially reduces but does not always eliminate gDNA. Even after digestion, roughly 1–2% residual gDNA can persist — which is why in-solution digestion with doubled enzyme units and post-digestion spin-column cleanup is recommended over on-column digestion alone for critical applications.
Does gDNA contamination affect RT-qPCR differently than RNA-seq?
In RT-qPCR, gDNA contamination produces signal in no-RT controls and overestimates expression, particularly for intronless genes such as some mitochondrial targets or when primers don't span introns. In RNA-seq, effects are most pronounced for low-abundance transcripts, with susceptibility varying significantly by library preparation method.
Which RNA-seq library preparation method is more susceptible to gDNA contamination?
Ribo-Zero depletion libraries capture significantly more gDNA than Poly(A) selection libraries because Ribo-Zero retains all non-rRNA sequences (including gDNA), while Poly(A) selection specifically targets transcripts with poly-A tails that gDNA structurally lacks. This makes prior DNase treatment critically more important when using Ribo-Zero protocols.


