
Introduction
Many clinical and research labs struggle with inconsistent DNA quality that compromises downstream test results. In molecular diagnostics, where patient care decisions hinge on accurate sequencing and PCR data, unreliable DNA extraction creates serious workflow bottlenecks and diagnostic uncertainty.
Automated genomic DNA extraction uses instruments and reagent systems to isolate intact, high-purity gDNA from whole blood without the variability of manual handling. For labs running NGS, PCR, or microarray workflows, that consistency is the difference between reliable results and repeated troubleshooting.
This guide covers what the process involves, why automation has become the standard, how each protocol step works, and which variables most affect outcomes.
Key Takeaways
- Automated gDNA extraction isolates high-purity genomic DNA through four core stages: lysis, binding, washing, and elution using magnetic bead technology
- Reduces hands-on time to approximately 3.3 minutes per sample and delivers reproducible purity ratios (A260/280 ~1.8, A260/230 ≥2.0)
- Whole blood sample quality, anticoagulant type, and incubation parameters determine extraction success
- Low yield, poor purity, and inhibitor carryover typically trace back to pre-analytical handling errors
- Modern systems like the Manta process single samples without batch minimums, removing pressure to accumulate runs
What Is Automated Genomic DNA Extraction from Whole Blood?
Automated gDNA extraction uses programmable instruments and pre-configured reagent systems to isolate genomic DNA from white blood cells (leukocytes) in whole blood. The process removes proteins, lipids, and other contaminants in a controlled, repeatable sequence.
The goal is a DNA eluate with sufficient yield (measured in micrograms), high purity (A260/280 of ~1.8, A260/230 of ≥1.6–2.0), and intact fragment length suitable for downstream applications. According to Thermo Fisher Scientific, an A260/280 ratio of ~1.8 is generally accepted as pure for DNA, while abnormal ratios indicate contamination by residual phenol, guanidine, or other extraction reagents.
Understanding what this process produces also means knowing what it isn't. Automated gDNA extraction from whole blood targets cellular DNA within leukocytes and requires complete cell lysis to release high-molecular-weight chromosomal DNA — a different objective than:
- RNA extraction, which needs distinct lysis conditions and RNase inhibitors to prevent degradation
- Cell-free DNA (cfDNA) extraction, which isolates short fragments (~167 bp) circulating freely in plasma, not cellular DNA from blood cells
Healthy blood contains 4–7 × 10⁶ leukocytes per milliliter, yielding 30–40 µg of DNA per mL. From a standard 200 µL input, labs can expect 4–12 µg of gDNA when the protocol runs correctly.
Why Clinical and Research Labs Use Automated gDNA Extraction
Reproducibility Across Operators
Manual extraction introduces operator-to-operator variability that compromises reproducibility. Automated magnetic bead methods achieve 11.8% coefficient of variation versus 16% for manual spin-column methods, delivering tighter reproducibility across operators and runs. This matters in clinical settings where diagnostic decisions depend on consistent DNA quality across batches and technicians.

Throughput and Lab Economics
Manual extraction requires approximately 6.9 minutes of hands-on time per sample versus 3.3 minutes for automated platforms. In a 30-sample batch, that difference frees up 78 minutes of operator time — time that shifts to data analysis and quality review instead. Cambrian Bioworks' Manta completes a run in approximately 28 minutes with less than 10 minutes of active handling, lowering cost per test by up to 56%. For labs processing more than 50 samples per day, these gains compound fast.
Contamination Risk Mitigation
Manual pipetting during lysis and wash stages increases the risk of:
- Cross-contamination between samples
- Aerosol hazards that compromise safety
- Sample-to-environment exposure
Automated closed-system designs mitigate this risk. Systems compatible with biosafety cabinets — including instruments compact enough to fit inside a hood — provide additional containment for hazardous or infectious samples.
Critical Application Settings
Automated extraction is now standard practice in:
- Clinical diagnostics, where reproducibility directly affects patient care decisions
- Biobanking facilities processing large sample volumes at consistent quality
- Oncology labs running somatic mutation profiling that requires high DNA integrity
- Transplant medicine centers performing HLA typing with zero contamination tolerance
- Pharmacogenomics labs detecting variants that demand inhibitor-free DNA
- Genetic disease screening programs relying on DNA suitable for sensitive PCR assays
NGS workflows require high integrity (DIN ≥7), while PCR-based assays tolerate moderate fragmentation but remain intolerant of inhibitors — a distinction that shapes both kit selection and protocol design.
Regulatory and Quality Expectations
Clinical-grade extraction workflows increasingly require IVD-certified reagents and instruments. CE-IVD certification under Regulation (EU) 2017/746 (IVDR) matters when extracted DNA feeds into regulated diagnostic pipelines. The Manta system holds CE-IVD certification, ensuring compliance with rigorous standards for in vitro diagnostic devices. In India, Manta also holds CDSCO IVD certification (licence number MFG/IVD/2025/000069).
Labs seeking ISO 15189 accreditation must demonstrate quality monitoring across all pre-analytical and analytical steps — a requirement that automated systems with documented reproducibility help satisfy.
How Automated gDNA Extraction from Whole Blood Works: The Step-by-Step Protocol
Automated gDNA extraction follows a consistent four-stage workflow:
- Whole blood is combined with lysis reagents and Proteinase K
- Lysate is mixed with magnetic beads in a binding buffer
- Beads with bound DNA are washed through sequential wash buffers
- DNA is eluted from the beads into a clean buffer
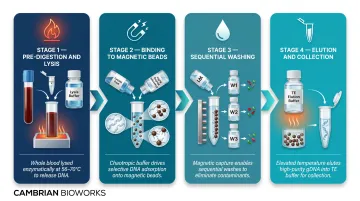
All steps are executed by an automated instrument that handles fluid transfers, mixing, temperature control, and magnetic bead manipulation. Each stage depends on precise reagent chemistry — here's what each component does.
Reagent Components
- Proteinase K — a broad-spectrum serine protease that degrades histones bound to DNA and inactivates RNases/DNases. Optimal activity: 55–65°C at 0.2–1 mg/mL.
- Blood Lysis Buffer — disrupts red and white blood cell membranes to release cellular contents.
- Binding Buffer — chaotropic salts destabilize hydrogen bonds and promote nucleic acid adsorption onto magnetic bead surfaces.
- Elution Buffer — typically Tris-EDTA (TE) buffer or nuclease-free water at pH 8–9, which releases purified DNA from the beads.
Step 1: Pre-Digestion and Lysis
Whole blood (typically 200–250 µL) is combined with Proteinase K and Blood Lysis Buffer in a microcentrifuge tube, vortexed thoroughly, then incubated at 56–70°C for 10–25 minutes depending on the protocol.
Why temperature matters: Too low risks incomplete protein digestion that leaves enzymes active to degrade DNA. Too high risks DNA denaturation. Hemolyzed or challenging samples may require additional lysis-stabilizing buffers.
Step 2: Binding to Magnetic Beads
The lysate is transferred to the binding well or combined with magnetic bead suspension. The binding buffer's chaotropic composition promotes DNA adsorption onto bead surfaces while the instrument mixes the sample to ensure uniform bead-DNA contact.
Magnetic bead recovery rates average 94.98%, with nucleic acid recovery at 91.83%, demonstrating high capture efficiency when bead-to-sample ratios and mixing dynamics are correctly calibrated.
Step 3: Sequential Washing
The automated system uses a magnetic array to capture and immobilize beads while the instrument aspirates supernatant. Wash buffers serve distinct, sequential purposes:
- Early washes — remove proteins and bulk cellular debris
- Middle washes — strip residual chaotropic salts from the bead surface
- Final washes — remove organic contaminants and any remaining inhibitors
Wash buffer composition, incubation time at each step, and bead-drying duration before elution all affect final purity ratios. Ethanol-based wash buffers with surfactants like Tween 20 are common. Typical drying time is 5–10 minutes to prevent bead cracking while ensuring efficient elution.
Step 4: Elution and Collection
Elution buffer is added directly onto the bead pellet. The magnet disengages, and the instrument mixes and incubates at elevated temperature (typically 65–80°C) to fully rehydrate and release DNA from the beads.
Expected output from 200 µL whole blood:
| Metric | Manual Methods | Automated (General) | Manta (Cambrian Bioworks) |
|---|---|---|---|
| Yield | 4–12 µg | 4–15 µg | 6–15 ng/µL |
| A260/280 | 1.7–1.9 | 1.7–1.9 | 1.8–2.0 |
| A260/230 | ≥2.0 | ≥2.0 | 2.0–2.2 |
Elution volume is adjustable: larger volumes increase total DNA recovery but reduce concentration; smaller volumes produce higher concentration at the cost of total yield. Eluted DNA is transferred to a collection tube and stored at −20°C.
Key Factors That Determine Extraction Quality
Sample Pre-Analytics
Anticoagulant type has direct downstream impact:
- EDTA tubes — preferred standard; preserve DNA without introducing PCR inhibitors
- Heparin tubes — inhibit Taq polymerase at 5-20 IU/mL, causing PCR failure; avoid for PCR-based applications
- Citrate tubes — acceptable but yield lower DNA due to dilution
Whole peripheral blood stored at room temperature for 8 days retains only 17.8% intact DNA; storage at +4°C preserves 66.7% after 8 days. Storage at -80°C reduces yield by 23% and amplifiability by 30% compared to fresh samples.
Freeze-thaw cycles cause severe damage: Ten freeze-thaw cycles reduced DNA yield by 47% and diminished the probability of achieving optimal A260/280 purity by 28%. Aliquot whole blood before freezing to avoid repeated freeze-thaw.

Lysis Efficiency
Incomplete lysis — due to insufficient Proteinase K, inadequate incubation time, or incorrect temperature — is the most common upstream failure point. Residual protein contamination carries through to the eluate, lowering A260/280 ratios and causing enzyme inhibition in PCR.
Hemolyzed samples require adjusted protocols. Documented impact by severity:
- Free hemoglobin at 1.6 µM causes complete fluorescence quenching of EvaGreen in qPCR
- 39 µM free hemoglobin significantly decreases positive reactions in dPCR
- Severe hemolysis (>4 g/L free hemoglobin) decreases NGS library concentration from 9.69 to 7.30 ng/µL and increases duplication rates
Reject visibly hemolyzed samples or implement hemolysis grading before extraction.
Bead Handling and Wash Stringency
Magnetic bead concentration, aspiration speed during supernatant removal, and drying time all require calibration to the specific instrument and reagent system. Over-washing strips weakly bound DNA and reduces yield; under-washing leaves inhibitors in the eluate.
Drying time is a narrow window: over-dried beads crack and fail to release DNA during elution, while residual ethanol from insufficient drying contaminates the eluate and degrades purity ratios.
Scale and Throughput Considerations
Extraction performance can vary between single-sample and batch runs if the instrument's fluid handling isn't optimised for each scale. Systems with minimum batch size requirements create queuing delays in clinical settings where sample volumes fluctuate throughout the day.
The Manta system addresses this directly — processing 1–32 samples per run, it allows labs to run extractions as samples arrive rather than holding them until a full batch accumulates.
DNA Integrity for Downstream Applications
A260/280 ratio — indicates protein contamination; acceptable range is 1.7-1.9 (target ~1.8)
A260/230 ratio — indicates salt/solvent contamination; acceptable range is ≥1.6-2.0 (target ~2.0)
DIN (DNA Integrity Number) — measured on a 1-10 scale where 1 = highly degraded and 10 = fully intact. Intact gDNA from healthy donor blood typically scores DIN 9.8.
Minimum thresholds by application:
| Application | A260/280 | A260/230 | DIN | Minimum Yield |
|---|---|---|---|---|
| PCR | 1.7-1.9 | ≥1.6 | Not critical | Variable |
| NGS | 1.8-2.0 | 2.0-2.2 | ≥7.0 | ≥500 ng |
| Microarray | 1.8-2.0 | ≥1.8 | Not critical | Variable |
GENEWIZ requires DIN >7.0 for whole genome sequencing submissions, along with A260/280 of 1.8-2.0 and minimum 100 ng DNA.
Common Issues, Mistakes, and Misconceptions
Misconception: Automation Guarantees Quality Regardless of Sample Condition
Automation standardizes the process — it cannot rescue degraded, improperly stored, or over-hemolyzed samples. Pre-analytical handling errors are the primary cause of extraction failure, not instrument malfunction. Prompt extraction and minimal biospecimen storage time matter more than which platform you run.
Mistake: Shortening the Lysis Incubation
Labs under throughput pressure sometimes reduce lysis time or temperature. The result: residual protein contamination that elevates background absorbance, skews A260/A280 ratios, and causes enzyme inhibition in downstream PCR. Proteinase K needs adequate time at 55–65°C to fully digest histones and nucleases — cutting this step short undermines every step that follows.
Misconception: All Automated Extractors Perform Equivalently
Real performance differences exist across platforms. Magnetic array strength, mixing dynamics, temperature control precision, and cartridge sealing each affect yield and purity — particularly with challenging inputs like hemolyzed, frozen, or low-volume samples. Validate the system against your intended sample types before clinical deployment.
Mistake: Ignoring Anticoagulant Type
Heparin tubes are incompatible with PCR-bound workflows. Heparin strongly inhibits Taq polymerase and cannot be fully removed during extraction — even trace carry-over will cause assay failure. Always specify EDTA tubes in your SOPs.
Quick Reference: Common Failures and Fixes
| Issue | Root Cause | Fix |
|---|---|---|
| Low yield / failed extraction | Degraded or hemolyzed sample | Enforce pre-analytical handling SOPs |
| Poor A260/A280 ratios | Shortened lysis incubation | Maintain full 55–65°C incubation time |
| Inconsistent results across runs | Unvalidated extraction platform | Validate against your specific sample types |
| PCR inhibition despite clean extraction | Heparin anticoagulant tubes used | Switch to EDTA collection tubes |

Frequently Asked Questions
What anticoagulant tube is best for automated genomic DNA extraction from whole blood?
EDTA tubes are the standard choice — they preserve DNA without introducing PCR inhibitors. Avoid heparin tubes for PCR-based applications, as heparin strongly inhibits Taq polymerase at 5-20 IU/mL. Citrate tubes are acceptable but yield slightly less DNA due to sample dilution.
Can I use frozen whole blood for automated gDNA extraction?
Frozen blood (EDTA, -80°C) is compatible with most automated protocols, though -80°C storage reduces yield by 23% and amplifiability by 30% versus fresh samples. Repeated freeze-thaw cycles shear DNA — limit to one cycle, thaw on ice, and process promptly.
What DNA purity ratios should automated whole blood extraction produce?
Target A260/280 of ~1.8 (acceptable: 1.7-1.9) and A260/230 of ~2.0 (acceptable: ≥1.6). Values outside these ranges indicate protein, salt, or solvent contamination that can inhibit downstream reactions. The Manta system consistently delivers A260/280 of 1.8-2.0 and A260/230 of 2.0-2.2.
How much whole blood is needed for a standard automated gDNA extraction?
Most automated protocols use 200-250 µL of whole blood per reaction, yielding 4-12 µg of gDNA — sufficient for multiple downstream analyses. Some protocols support up to 2 mL input for biobanking or large-scale applications. The Manta system accommodates input volumes from 200 µL to 2 mL.
How do I know if my extracted gDNA is suitable for NGS?
For NGS, your sample needs A260/280 ~1.8, A260/230 ~2.0, yield ≥500 ng total, and DIN ≥7. Confirm purity by spectrophotometry and DIN by TapeStation or gel electrophoresis — most NGS service providers require DIN >7 before accepting samples for library preparation.
What causes low DNA yield in automated whole blood extraction?
Low yield typically traces back to one of four root causes: insufficient lysis (low Proteinase K activity or short incubation), poor bead binding from incorrect buffer ratios, over-stringent washes stripping bound DNA, or a degraded input sample from improper storage. Pre-analytical handling errors account for the majority of failures.


